|
|
|
L’anatomie et les fonctions des cellules Le sens de l’information génique (II) Pour transférer une information, réparer une blessure ou copier une chaîne spécifique de molécules, tous les organismes vivants ont besoin des deux acides nucléiques, l'ARN et l'ADN, qui seront tantôt viraux ou cellulaires en fonction du sujet. Grâce au code génétique, la cellule eucaryote est capable de synthétiser les protéines qui véhiculeront les besoins vitaux à travers tout l'organisme (voir plus bas). Ces protéines sont elles-mêmes assistées par d'autres protéines qui favorisent les réactions chimiques, ce sont les enzymes. Mais les processus biologiques ne se limitent pas à ces échanges car la biologie moléculaire est plus ambitieuse. Thomas Cech[3] de l'Université de Californie et indépendamment Sidney Altman de l’Université de Yale découvrirent en 1983 qu'un protozoaire thermophile disposait d'un ARN ribosomial qui fonctionnait comme une enzyme : ils baptisèrent cette entité un "ribosyme". Or jusqu'alors les acides aminés n'étaient pas considérés comme des protéines. La découverte de Cech et de ses collaborateurs confirmerait l'idée selon laquelle les premiers êtres vivants seraient des ARN ribosomiques ou du moins à fonction catalytique capables de se reproduire seuls, bien que leur séquence de nucléotides doive être préservée d’une manière qui demeure encore mystérieuse.
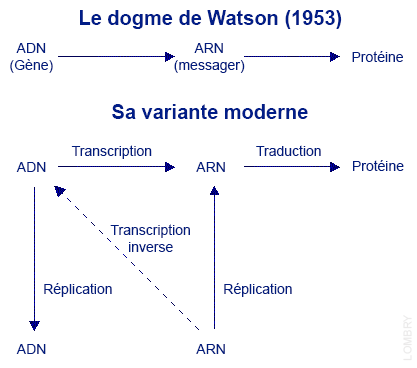
Nous verrons à propos de la chimie prébiotique que des expériences ont montré que des molécules complexes peuvent se polymériser sur des surfaces argileuses et même de l'ARN peut se former dans un atmosphère sous certaines conditions. En 2022, des chercheurs ont également montré que des ARN peuvent muter et se développer en suivant l'évolution drawinienne. Trois arguments viennent renforcer le rôle essentiel de l'ARN : 1°. Un polymère d’une variété de nucléotides peut contenir une information génétique (comme le fait l’ADN) 2°. Il existe des preuves expérimentales que la polymérisation de l’HCN (ou l'hydrolyse des polycyanogènes) peut conduire à la formation des purines (des tautomères de forme CnH et NnH), y compris celles constituants les molécules d'ARN modernes, l’adénine (forme brute C5H5N5) et la guanine (C5H5N5O). 3°. Des réactions chimiques simples liant les nucléotides sont omniprésentes dans tous les organismes, sous forme de coenzymes ou d’ATP. En découvrant la double hélice de l’ADN, Watson et Crick précisaient le sens de la transmission de l'information génique : stockée dans les gènes de l’ADN, cette information est transcrite par l'ARN messager qui la traduit sous forme de protéine. En 1970, David Baltimore de MIT, Satoshi Mazutani et Howard Temin de l’Université du Wisconsin découvrirent que dans certains virus, tel le sarcome de Rous (tumeur) ou le FeL V (leucémie féline), une enzyme comme la transcriptase inverse renversait le sens de l’information génique : l'ARN viral fait office de code génétique et c'est la transcriptase inverse qui retranscrit l'ADN. Le noyau cellulaire est alors infecté. La cellule subit une mutation de son génome qui active le développement d'un cancer. Si l'ADN abrite un oncogène, la cellule dégénérera et deviendra cancéreuse.
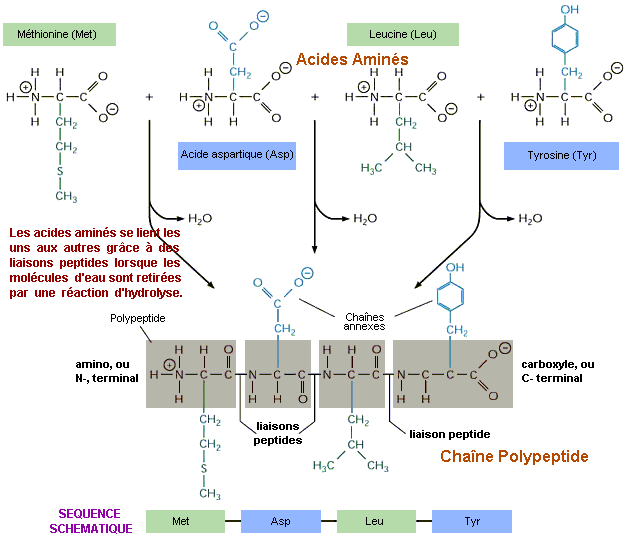
La découverte de la transcription inverse ou rétro-transcription explique bon nombre de maladies résistantes, en particulier le fait qu'elles peuvent se transmettre à des animaux non consanguins et que certaines tumeurs provoquent une déficience des protections immunitaires de l'organisme. Ces rétrovirus seront malheureusement découverts chez l’être humain en 1978. Le VIH vecteur du SIDA et le SARS-CoV-2 vecteur de la Covid-19 comptent parmi ceux-ci. Enfin, les protéines qu'on retrouve dans les acides nucléiques (hétéroprotéines) ou les tissus (holoprotéines) sont incapables de se reproduire. Les anticorps par exemple qui assurent notre défense immunitaire sont une variété de protéines synthétisées par les globules blancs. Une fois libérés dans le sang, leur tâche consiste à neutraliser les antigènes, un point c'est tout. Les protéines doivent donc être copiées. Cette technique est vieille de plus de 3.8 milliards d'années. La synthèse des protéines Nous avons expliqué brièvement (voir page 1) que la cellule fabrique des acides aminés grâce à l'expression des gènes. Ces acides aminés représentent les briques des protéines qui assurent les fonctions métaboliques de l'organisme. On estime que les quelque 19969 gènes (leur nombre exact reste incertain, cf. P.Amaral et al., 2023) expriment entre 80000 et 400000 protéines différentes dont l'identification est toujours en cours dans le cadre de divers projets dont l'initiative HUPO (Human Proteome Organization) lancée en 2001. Cependant, toutes ces protéines qui forment le protéome ne sont pas toutes produites par toutes les cellules du corps au même moment car les cellules ont des protéomes différents selon leur type cellulaire. Comment la cellule fabrique-t-elle ces acides aminés et ces protéines ? Comment la cellule communique-t-elle avec son environnement ? Dans son principe, quand une cellule eucaryote a besoin d'une substance spécifique pour assurer ses fonctions métaboliques, Francis Crick a découvert à la fin des années 1950 que l'information se transmettait essentiellement de l'ADN vers l'ARN aux protéines au cours d'un processus qui se déroule en plusieurs étapes centrées sur une transcription et une traduction de l'information au cours de laquelle la molécule d'ADN est fractionnée (épissage). On y reviendra. Les deux brins de la double hélice de l’ADN formés de bases complémentaires constituent en quelque sorte un original et sa copie. A partir de l'ADN du noyau, un gène est transcrit sous forme d'ARN prémessager (pre-ARNm). L’ADN se divise sur une certaine longueur sous l’action d’un complexe multienzymatique, le réplisome. Contrairement à la représentation souvent donnée, selon Christian de Duve le réplisome est fixe et probablement ancré au niveau du mésosome et c’est l’ADN qui se déplace. Il se présente symétriquement à travers deux réplisomes jumeaux et sort du complexe sous forme dupliquée. Ce mécanisme est très lourd car il doit surmonter de nombreux problèmes topologiques et mécaniques liés à la structure hélicoïdale et l’orientation antiparallèle des deux brins d’ADN, et veiller à ce que la réaction enzymatique soit amorcée par un ARN. Ca c'est en théorie car en pratique le processus est bien plus complexe et n'est pas encore totalement élucidé. A
voir : Le
génome, comment ça marche ?,
Inserm L'ADN et l'ARNm - De l'ARN à la protéine, F.Blasselle ADN et synthèse d'une protéine, S.Sahlaoui Translation : Synthesis of a protein, NDSU Virtualo Cell
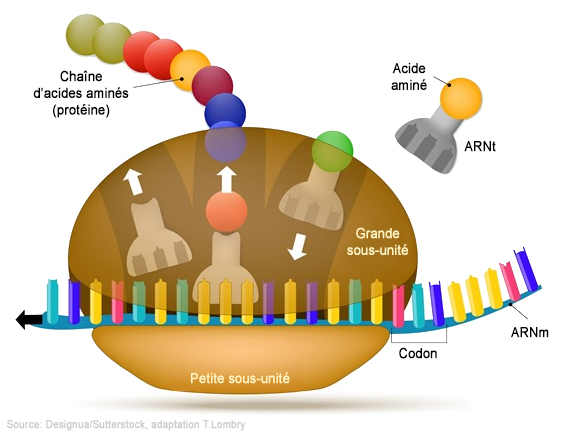
Au cours de l'expression des gènes, la transcription de l'ADN a également l'immense tâche de coder pour les ARN messagers (ARNm), c’est-à-dire de synthétiser les protéines. Noter que pour une cellule nerveuse on ne parle pas de messager mais de neurotransmetteur, telle la dopamine qui est une molécule spécifique aux neurones. Comme l'explique le schéma présenté ci-dessus à droite, lorsque la copie est effectuée, l’ARNm va transporter son information dans le cytoplasme où elle sera lue par l’ARNr. Malgré trois décades d’investigations, il n’y a que des hypothèses sur le fonctionnement des ribosomes, eux-mêmes assemblés à partir d’ARN spécifiques et de protéines. Leur fonction est de construire des protéines à partir des acides aminés. Mais ces derniers sont incapables de lire le message de l’ARNm. Ils doivent faire appel aux ARNt qui capturent les acides aminés par attraction chimique. Lorsqu’un message se présente dans (ou peut-être sur) la petite usine chimique du ribosome, les deux sous-unités ribosomiales se rapprochent et le message en question est dirigé vers la chaîne de montage. A l’extrémité de l’ARN de transfert se trouve des anticodons (combinaisons de 3 bases), complémentaires des codons du message. L’acide aminé les reconnaît, se détache de l’ARN de transfert et s’accroche à la chaîne polypeptidique en construction (cf. schéma). La protéine, copie fidèle de la séquence de l’ADN se construit codon par codon au rythme d’une vingtaine par seconde. Une fois libéré de son message, l’ARN de transfert retourne capturer un nouvel acide aminé. Lorsque la synthèse est terminée, les deux sous-unités ribosomiales se séparent, la protéine se libère et se replie sur elle-même en prenant la forme correspondant à ses affinités chimiques. Dès cet instant la protéine constituée d'acides aminés est active. Elle peut par exemple interagir avec le noyau pour interrompre l'activité nucléique, synthétiser des substances antidouleurs, etc.
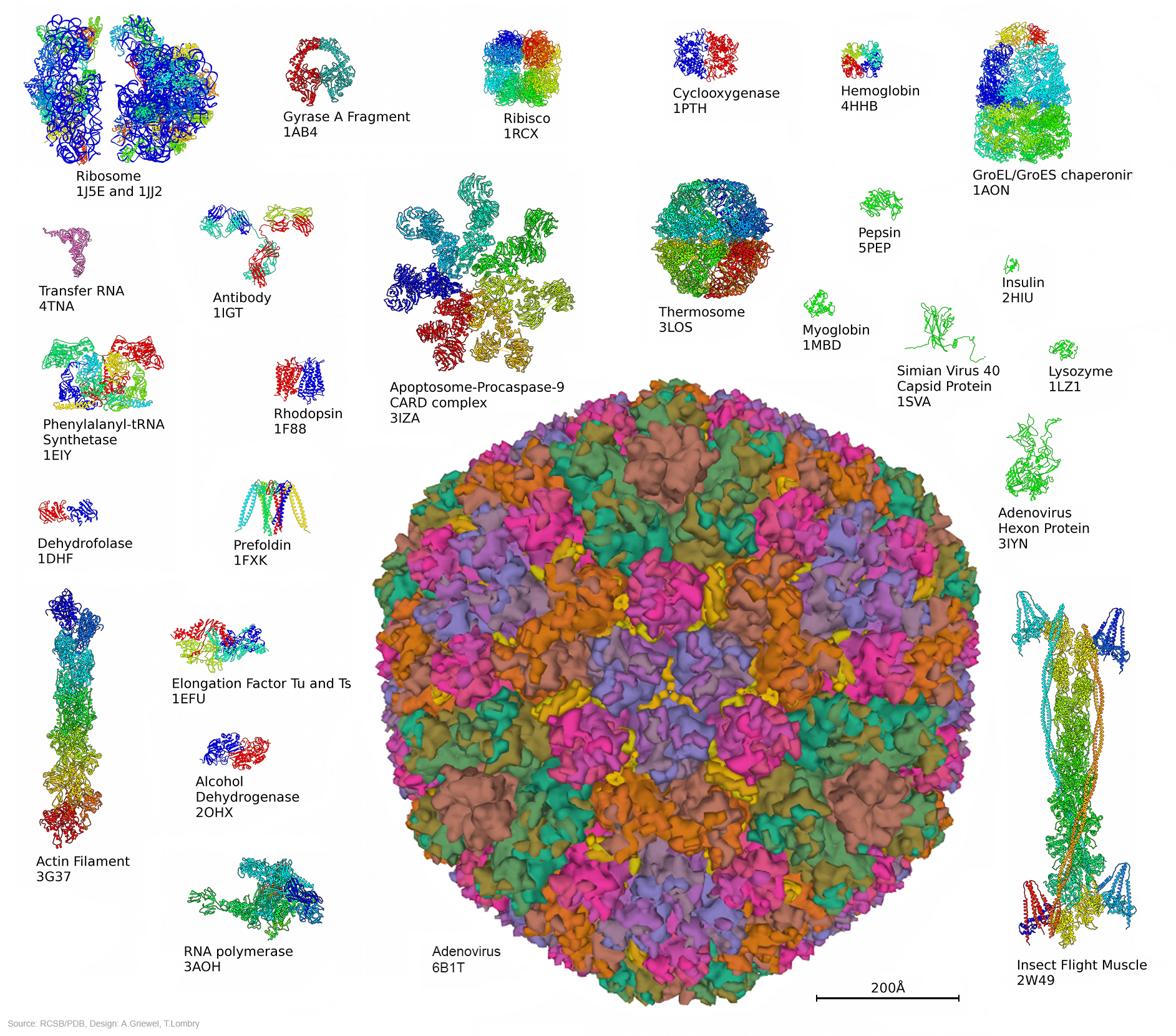
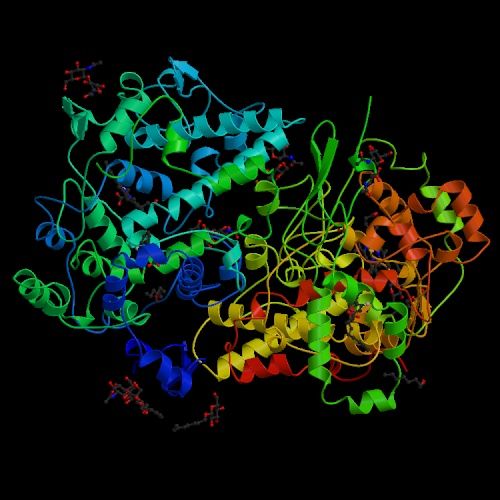
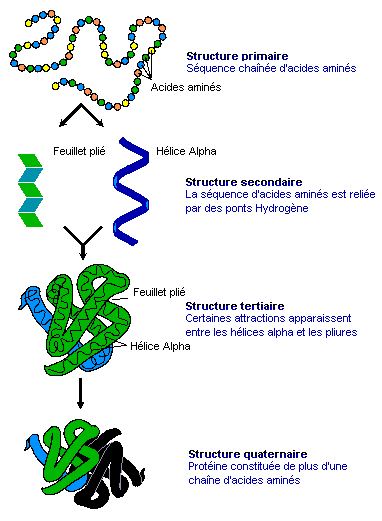
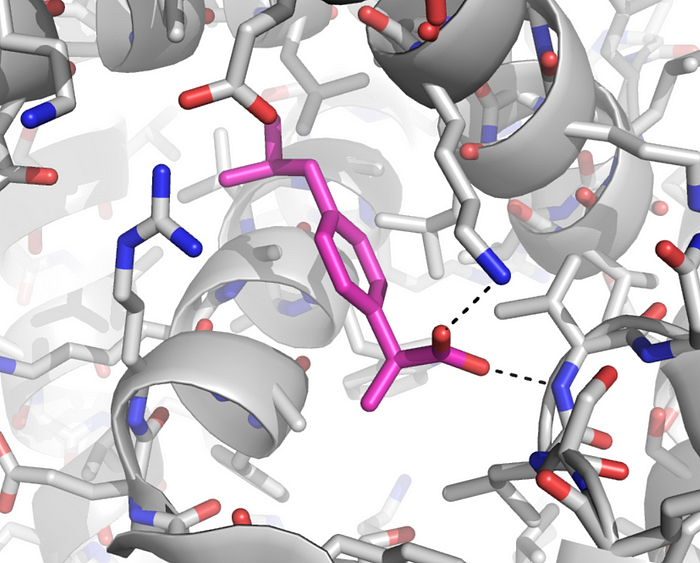
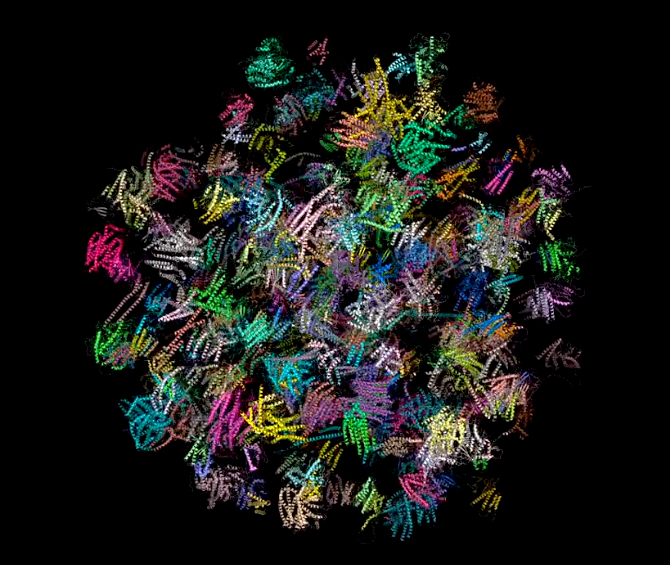
Chacun connaît par exemple les bienfaits de l’aspirine. Pourquoi agit-elle ? Lorsqu’il y a un dérèglement métabolique, nous venons de voir que les cellules envoient des messages chimiques sous forme de molécules. Parmi ces messages, elles peuvent demander la synthèse de prostaglandine afin de provoquer la fièvre, une inflammation ou manifester des douleurs. Elles peuvent également synthétiser de la thromboxane pour former des caillots ou de la prostacycline lors d’une irritation de l’estomac par exemple. C’est pour supprimer ces malaises ou plus précisément pour bloquer les trois types de messages à l’origine de ces symptômes que l’aspirine fut inventée. Si les molécules de protéines diffèrent naturellement les unes des autres par leur séquence d’acides aminés, leurs fonctions diffèrent crucialement de leur forme. Il subsiste une grande incertitude à propos du mécanisme qui donne leur forme aux molécules; pour toutes les molécules sauf les plus petites, il semble y avoir un grand nombre d’étapes de pliures intermédiaires qui demeurent inactives. Une explication serait de considérer que le pliage est partiellement déterminé par l’ordre dans lequel les acides aminés sont traduits dans les ribosomes, mais il existe des preuves que d’autres protéines (les chaperons) assistent à ce processus. La réponse est incertaine et il pourrait ne pas y avoir de règle générale. Paradoxalement, la synthèse des acides nucléiques (les bases de l'ADN) n'est possible qu'en présences d'enzymes, de protéines qui assurent une fonction de catalyseur. Ces protéines sont essentielles car elles constituent la cheville ouvrière de la cellule, non seulement elles créent les enzymes mais elles forment les parois, les anticorps, etc. Il est donc difficile de savoir qui de l'ADN ou de l'ARN est le premier. Si la plupart des ARN fonctionnels codent les protéines, certains ARN ne codent pas de protéines mais assurent d'autres fonctions. Ainsi, nous avons vu que les ARN ribosomaux (ARNr) et les ARN de transfert (ARNt) interviennent essentiellement durant le processus de traduction de l'ARNm. L'ARN nucléaire (pARNn) contrôle l'épissage des ARNm et les microARN (miARN) bloquent la traduction des ARNm en s'appariant avec leurs séquences complémentaires. Enfin, la synthèse des protéines rejette de l'azote qui devient une substance toxique pour l'organisme à forte concentration. La cellule doit donc s'en débarrasser. Grâce à l'action des mitochondries, l'azote et l'ammonium (ion NH4+) issu du métabolisme cellulaire sont transformés en urée qui est évacuée dans le cytoplasme. Elle est ensuite récupérée par le système circulatoire jusqu'aux reins, où le sang est filtré et débarrassé de l'urée sous forme d'urine puis expulsée du corps. Notons que cette urée contenant de l'azote, elle peut servir d'engrais naturel. De même que l'urine qui comprend également une grande quantité de phosphore dont la concentration dépend des habitudes alimentaires. Le phosphore est un élément très précieux qu'on extrait normalement à grand prix de roches phosphatées. Nous avons tord de le jeter à l'égoût. En effet, cet élément est très recherché car sous forme de phosphate ou d'acide phosphorique il entre dans la composition de nombreux produits : engrais, stabilisant, pâte dentrifrice, détartrant, additif alimentaire (E 338), agent anticorrosif , etc. La structure des protéines Nous connaissons environ 200 millions de protéines mais nous connaissons seulement la structure spatiale ou 3D d'environ 20% d'entre elles. Pourquoi la structure 3D d'une protéine est-elle importante ? Comme une prise mâle spécifique s'emboîte dans une prise femelle adaptée, la forme d'une protéine nous renseigne sur sa fonction. Comparons une protéine avec une main. En posant une main à plat ou sur base de sa seule silhouette, il est impossible d'imaginer sa complexité et savoir comment elle fonctionne. En revanche, présentée sur une radiographie 3D, les différents degrés de libertés du poignet et des doigts apparaissent, permettant de comprendre comme s'effectue la préhension des objets. Le même principe s'applique à l'étude des protéines et des acides aminés qu'elles portent. Connaître la structure 3D des protéines est donc essentielle pour connaître son orientation dans l'espace, sa dynamique, les acides aminés présents sur sa surface, comprendre leurs mécanismes et surtout la manière dont la protéine s'agence avec d'autres molécules et sa compatibilité. Ces protéines peuvent également avoir un intérêt thérapeutique. Trois grandes méthodes permettent de caractériser la structure 3D des protéines : la résonance magnétique nucléaire (RMN), la cristallographie (la diffraction aux rayons X) et la cryomicroscopie électronique (cryo-ME) dont les données sont ensuite représentées graphiquement sur ordinateur. Pour représenter les moléculaires et leur activité biologique, les biologistes moléculaires ont défini quatre types de structures protéiques : La structure primaire est la simple séquence illustrant l'enchaînement des acides aminés. Elle est intéressante pour décrire une molécule mais insuffisante pour définir une activité enzymatique. Par exemple, un peptide présentant une séquence d'acides aminés particulière présente une activité biologique. Si on lui ajoute un peu de détergent on perturbe sa structure spatiale et on détruit son activité biologique, phénomène qui ne peut pas apparaître dans ce schéma.
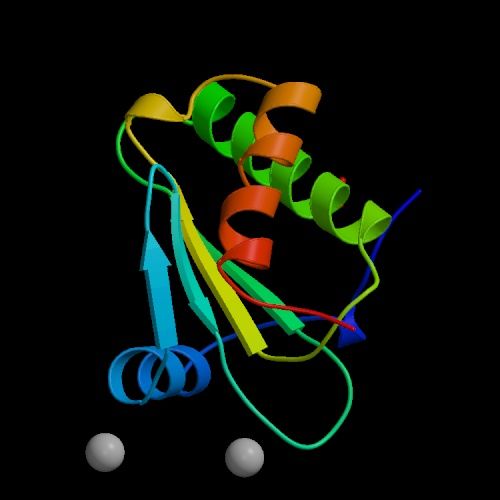
Documents adaptés de Genomics. La structure secondaire est un premier niveau d'agencement dans l'espace tenant compte des liaisons hydrogène. Elle met généralement en évidence certaines suites d'acides aminés qui se replient sous forme d'hélice alpha ou de feuillet bêta comme indiqué dans le schéma. La structure tertiair est un second niveau de structure tridimensionnel plus global insistant sur l'agencement des structures secondaires entre elles. Celles-ci forment un ensemble compact qui permet de définir les sites actifs pour les enzymes. La structure quaternaire est l'assemblage de plusieurs sous-unités distinctes (dimères, trimères, tétramères, etc), formant un ensemble tel qu'ils existe réellement in vivo. Cette représentation ne s'applique donc pas aux monomères Les "tire-bouchons" figurant sur l'image de droite représentant la ribonucléase P sont des hélices. Il en existe de plusieurs natures mais globalement elles conservent cette forme. Elle n'est pas aléatoire, elle est déterminée par des interactions internes de types hydrophobes. Les tracés plus fins en "fils de fer" sont des structures plus simples de types feuillets (bêta le plus souvent) et ou coudes (pour passer d'un feuillet à un autre ou vers une hélice,...). Ces représentations sont obtenues par modélisation par l'une trois méthodes précitées et par calculs mathématiques. Ils sont réalisés dans l'air ou dans l'eau et négligent de nombreux paramètres (force hydrophobe, sels, interactions avec le milieu, ....). De plus, il s'agit d'une image statique alors qu'en réalité la molécule est une structure dynamique. C'est un peu comme si on étudiait une main à plat sans se rendre compte que ses doigts peuvent bouger; cela ne reflète pas sa réalité physique ni ses fonctions. De nos jours, grâce à l'informatique, ces représentations se sont améliorées et dans les meilleurs cas les protéines et les acides aminés sont mis en contexte, au contact d'autres molécules d'intérêt (voir ci-dessous). De l'utilité des ordinateurs et de l'IA L'effet d'une protéine dépend de sa structure de l'échelle atomique à l'échelle moléculaire. Elle est donc très sensible, très complexe, qui plus est dynamique, et de ce fait elle est très difficile à modéliser dans l'espace. C'est la raison pour laquelle les chercheurs les étudient à l'aide de puissants ordinateurs et, de nos jours, en faisant appel à l'intelligence artificielle. Pourquoi a-t-on besoin d'une telle puissance de calcul ? Prenons un exemple. Une protéine est construite à partir de 20 acides aminés standards. Une seule protéine peut contenir aussi bien un seul acide aminé, quelques centaines voire plus de 2000 acides aminés. De ce fait la combinatoire devient vite astronomique. Comme il faut pouvoir prédire la structure 3D d'une protéine et la zone de compatibilté avec d'autres molécules (ainsi que d'autres paramètres comme sa biocomptabilité, sa solubilité, sa toxicité, etc), il faut déterminer dans l'espace la position exacte de chaque acide aminé et des atomes qu'elle porte. Dans une protéine contenant un seul acide aminé, il y a un choix parmi 20 séquences possibles. S'il y a 2 acides aminés, il y a 20x20 soit 400 séquences possibles. S'il y a 10 ou 100 acides aminés, les séquences possibles se comptent par milliards et sont ingérables sans ordinateur, sans même parler des séquences homologues qu'on retrouve chez d'autres organismes qui peuvent aider à construire la séquence protéique humaine. De plus, quand on développe de nouveaux médicaments, s'ajoute à cette combinatoire le fait qu'il faut calculer les interactions atomiques entre toutes les molécules pour déterminer quelle structure protéique est la plus adaptée à la solution thérapeutique. Dans le modèle "All atoms" d'AlphaFold de Google DeepMind, il faut pouvoir prédire les propriétés de l'interface entre l'acide aminé et les molécules d'intérêt. Concrètement, s'il y a un million de composés chimiques à tester contre la molécule (protéine) en développement, il y a un million de prédictions de structures à calculer ainsi que toutes les interactions protéines-ADN, etc, et toutes les modifications chimiques éventuelles que subissent les acides aminés. Compte tenu des Big data à gérer, ces calculs durent des mois et ne sont réalisables que sur les superordinateurs les plus puissants, généralement interconnectés (réseaux neuronaux, cloud et grid), ce qui explique que seules les plus grandes sociétés comme Google et des organisations scientifiques internationales ont les ressources (budget, infrastructure et compétences) nécessaires pour relever le défi. A consulter : HUPO - Protein Data Bank - UniProt AlphaFold Protein Structure Database
En 2021, les chercheurs de Google DeepMind et ceux du Laboratoire Européen de Biologie Moléculaire (EMBL) ont publié en libre accès les données de 20000 protéines exprimées par le génome humain. Cette base de données appelée AlphaFold Protein Structure Database (ou simplement AlphaFold) est complétée par 350000 protéines de 20 organismes, comprenant des bactéries et des souris, souvent utilisés par la recherche. Cette base de données contient un outil de recherche épaulé par l'intelligence artificielle qui permet de prédire avec précision la structure des protéines à partir d'une IA générative et de méthodes d'apprentissage profond (cf. J.Jumper et al., 2021). Pour ne pas surcharger cet article, nous décrirons ces concepts dans l'article consacré aux Avantages et inconvénients de l'intelligence artificielle. La dernière version d'AlphaFold (version 3 prévue en 2024) pourra prédire plusieurs formes 3D des protéines comme une version courte et une version longue (cf. l'épissage alternatif). Parmi les développements futurs, AlphaFold pourra également être mise en contexte et prédire les interactions des protéines avec d'autres molécules d'intérêt (modèle All atoms), c'est-à-dire faire des prédictions lorsque la protéine est liée à d'autres molécules (l'ADN, l'ARN, d'autres protéines, d'autres molécules, etc). En fait, cet outil est en développement permanent et comme le confirme le blog de DeepMind, chaque mois apporte son lot de nouveautés. Depuis, grâce à l'amélioration des performances des ordinateurs et en particulier des cartes GPU (graphiques), il existe des IA concurrentes comme EVO qui permet de travailler non seulement en "modalité unique", c'est-à-dire uniquement sur les protéines ou sur l'ARN mais également de réaliser des opérations multimodales sur une ou plusieurs molécules (protéines et ARNnc) et sur des séquences à l'échelle de génomes entiers. Selon EVO, les calculs sur des séquences partielles (jusqu'à 650 k de longueur par GPU) peuvent être effectués sur des stations graphiques ou des ordinateurs de bureau de dernière génération. Prochain chapitre
|
||||||||||||||||||||||||||||||||||||||||